
")
")
Sindromul Rett
Sindromul Rett este o tulburare genetică rară neurologică și de dezvoltare care este diagnosticată în perioada copilăriei. Aceasta afectează modul în care se dezvoltă creierul, cauzând o pierdere progresivă a abilităților motorii și a vorbirii. Această tulburare afectează în principal fetele.
Un copil care va avea sindrom Rett se va dezvolta normal în viaţă intrauterină. Majoritatea bebelușilor cu sindromul Rett par să se dezvolte normal în primele 6 până la 18 luni, apoi își pierd abilitățile pe care le aveau anterior - cum ar fi capacitatea de a se târî, de a merge, de a comunica sau de a-și folosi mâinile. Deși este prezent la naștere, sindromul Rett este încă nedetectat până la vârsta de aproximativ un an, atunci când copii îşi pierd majoritatea abilităților dobândite.
În timp, copiii cu sindromul Rett au din ce în ce mai multe probleme cu utilizarea mușchilor care controlează mișcarea, coordonarea și comunicarea. Sindromul Rett poate provoca, de asemenea, convulsii și dizabilități intelectuale. Mișcările anormale ale mâinilor, cum ar fi frecarea repetitivă sau bătăile din palme, înlocuiesc utilizarea intenționată a mâinilor.
Deși nu există un leac pentru sindromul Rett, se studiază potențiale tratamente. Tratamentul actual se concentrează pe îmbunătățirea mișcărilor și a comunicării, pe tratarea convulsiilor și pe oferirea de îngrijire și sprijin pentru copiii și adulții cu sindrom Rett și pentru familiile acestora.
Simptome
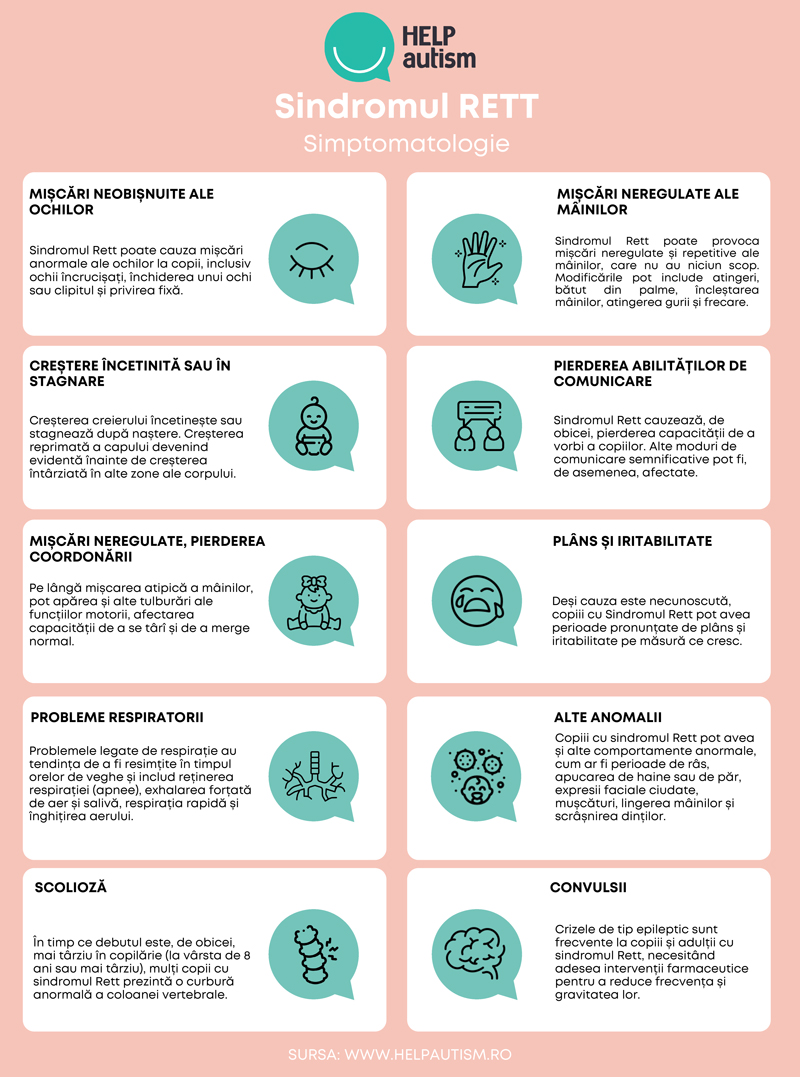
Semnele și simptomele sindromului Rett includ:
- Creștere încetinită. Creșterea creierului încetinește după naștere. Capul mai mic decât dimensiunea normală (microcefalie) este, de obicei, primul semn că un copil are sindromul Rett. Pe măsură ce copiii cresc, creșterea întârziată în alte părți ale corpului devine evidentă.
- Pierderea mișcărilor și a coordonării normale. Primele semne includ adesea un control redus al mâinilor și o capacitate tot mai redusă de a se târî sau de a merge normal. La început, această pierdere a abilităților se produce rapid, apoi continuă mai treptat. În cele din urmă, mușchii devin slabi sau pot deveni rigizi sau spastici, cu mișcări și poziții anormale.
- Pierderea capacității de comunicare. Copiii cu sindromul Rett încep, de obicei, să își piardă capacitatea de a vorbi, de a avea contact vizual și de a comunica în alte moduri. Ei pot deveni dezinteresați de alte persoane, de jucării și de mediul înconjurător. Unii copii au schimbări rapide, cum ar fi o pierdere bruscă a vorbirii. În timp, copiii pot recăpăta treptat contactul vizual și pot dezvolta abilități de comunicare nonverbală.
- Mișcări anormale ale mâinilor. Copiii cu sindromul Rett dezvoltă în mod obișnuit mișcări repetitive și fără scop ale mâinilor, care pot fi diferite pentru fiecare persoană. Mișcările mâinilor pot include fluturatul mâinilor, strângerea mâinilor, bătăile din palme, lovirea sau frecarea.
- Mișcări neobișnuite ale ochilor. Copiii cu sindromul Rett au tendința de a avea mișcări neobișnuite ale ochilor, cum ar fi privirea fixă intensă, clipitul, ochii încrucișați sau închiderea a câte un ochi.
- Probleme de respirație. Printre acestea se numără reținerea respirației, respirația anormal de rapidă (hiperventilație), exhalarea forțată a aerului sau a salivei și înghițirea aerului. Aceste probleme tind să apară în timpul orelor de veghe, dar tulburări de respirație, cum ar fi respirația superficială sau respirația periodică, pot apărea în timpul somnului.
- Iritabilitate și plâns. Copiii cu sindromul Rett pot deveni din ce în ce mai agitați și iritabili pe măsură ce cresc. Perioadele de plâns sau țipete pot începe brusc, fără un motiv aparent, și pot dura ore întregi. Unii copii pot experimenta temeri și anxietate.
- Alte comportamente anormale. Acestea pot include, de exemplu, expresii faciale ciudate și momente lungi de râs, lingerea mâinilor și apucarea părului sau a hainelor.
- Deficiențe cognitive. Pierderea abilităților poate fi însoțită de o pierdere a funcționării intelectuale.
- Convulsiile. Majoritatea persoanelor care au sindromul Rett suferă de convulsii la un moment dat în timpul vieții lor. Pot apărea mai multe tipuri de crize și sunt însoțite de o electroencefalogramă (EEG) anormală.
- Curbura anormală a coloanei vertebrale (scolioză). Scolioza este frecventă în cazul sindromului Rett. De obicei, începe între 8 și 11 ani și crește odată cu vârsta. Poate fi necesară o intervenție chirurgicală dacă curbura este severă.
- Bătăi neregulate ale inimii. Aceasta este o problemă care pune în pericol viața multor copii și adulți cu sindromul Rett și poate duce la moarte subită.
- Tulburări de somn. Tiparele anormale de somn pot include ore de somn neregulate, adormirea în timpul zilei și trezirea pe timp de noapte sau trezirea în timpul nopții cu plâns sau țipete.
- Alte simptome. Pot apărea o varietate de alte simptome, cum ar fi oase subțiri și fragile, predispuse la fracturi; mâini și picioare mici, de obicei reci; probleme de mestecat și de înghițire; probleme cu funcția intestinală; și scrâșnirea dinților.
Sindromul Rett este de obicei împărțit în patru stadii:
Stadiul I: Debut precoce, 6-18 luni.
Semnele și simptomele sunt subtile și ușor de trecut cu vederea în timpul primei etape, care începe între 6 și 18 luni și poate dura câteva luni sau un an. Bebelușii din acest stadiu pot arăta mai puțin contact vizual și încep să își piardă interesul pentru jucării. De asemenea, pot avea întârzieri în a se așeza sau a se târî.
Stadiul II: Deteriorare rapidă, 1-4 ani.
Începând cu vârsta cuprinsă între 1 și 4 ani, copiii pot pierde abilitățile pe care le aveau anterior. Această pierdere poate fi rapidă sau treptată, apărând pe parcursul a săptămâni sau luni. Apar simptomele sindromului Rett, cum ar fi creșterea încetinită a capului, mișcări anormale ale mâinilor, hiperventilație, țipete sau plâns fără motiv aparent, probleme de mișcare și coordonare și pierderea interacțiunii sociale și a comunicării. Tulburările de comunicare din sindromul Rett devin complexe în acest stadiu. Apar anomaliile respiratorii la 75% dintre copii, cu aspect de joc stereotip cu propria respirație. Aceste anomalii respiratorii se datorează unor disfuncții corticale care au conexiuni cu căile respiratorii Anomaliile EEG sunt prezente în toate cazurile, cu aspect de paroxisme de vârfuri sau ritm în regiunile centrale, pe un fond lent de unde delta şi teta.
Stadiul III: Platou, 2-10 ani.
Cea de-a treia etapă începe de obicei între 2 și 10 ani și poate dura mai mulți ani. Deși problemele cu mișcarea continuă, comportamentul poate avea o îmbunătățire limitată, cu mai puțin plâns și iritabilitate și o oarecare îmbunătățire a utilizării mâinilor și a comunicării. Convulsiile pot începe în această etapă și, în general, nu apar înainte de vârsta de 2 ani.
Stadiul IV: deteriorare motorie tardivă, 10 ani.
Acest stadiu începe de obicei după vârsta de 10 ani și poate dura ani sau decenii. Este marcat de mobilitate redusă, slăbiciune musculară, contracturi articulare și scolioză. Înțelegerea, comunicarea și abilitățile manuale rămân în general stabile sau se îmbunătățesc ușor, iar crizele pot apărea mai rar.
În versiunea anterioară a Manualului de Diagnostic si Clasificare Statistica a Tulburărilor Mintale, DSM-IV (publicat în 1994), sindromul Rett a fost clasificat ca fiind una dintre tulburările din spectrul autist (TSA), celelalte fiind tulburarea de spectru autist, sindromul Asperger, tulburarea de dezvoltare generalizată nespecificată și tulburarea dezintegrativă a copilăriei.
DSM-5, care a apărut în mai 2013, nu mai include sindromul Rett în mod specific ca fiind o tulburare de spectru autist. Utilizând această ediție, sindroamele specifice, cum ar fi sindromul Rett, care au caracteristici specifice autismului, nu sunt grupate sub noul diagnostic general de tulburare de spectru autist, ci sunt clasificate prin codul lor de diagnostic specific, care există deja pentru majoritatea sindroamelor genetice.
Deși este posibil ca sindromul Rett să nu fie în mod oficial un o tulburare de spectru autist în DSM-5, legătura cu autismul rămâne. Mulți copii sunt diagnosticați cu TSA înainte ca mutația MECP2 să fie identificată și apoi diagnosticul este revizuit în Rett.
Se estimează că sindromul Rett afectează una la fiecare 10.000-15.000 de fete născute vii din toate grupurile rasiale și etnice din întreaga lume. Testarea prenatală este disponibilă pentru familiile cu o fiică afectată care are o mutație MECP2 identificată. Cu toate acestea, deoarece tulburarea apare spontan la majoritatea persoanelor afectate, riscul ca o familie să aibă un al doilea copil cu această tulburare este mai mic de 1%.
Povestea este diferită în cazul băieților care au o mutație MECP2 cunoscută ca fiind cauza sindromului Rett la fete. Deoarece băieții au doar un singur cromozom X (și un cromozom Y), le lipsește o copie de rezervă care ar putea să compenseze pentru cel defect și nu au nicio protecție împotriva efectelor dăunătoare ale tulburării. Băieții cu un astfel de defect frecvent nu prezintă trăsături clinice ale sindromului Rett, dar se confruntă cu probleme grave atunci când se nasc și mor la scurt timp după naștere. Un număr foarte mic de băieți pot avea o mutație diferită în gena MECP2 sau o mutație sporadică după concepție, care poate cauza un anumit grad de dizabilitate intelectuală și probleme de dezvoltare.
Diagnostic diferențial se impune cu toate celelalte tulburări pervazive de dezvoltare, faţă de care se diferențiază prin existența progredienței simptomatologiei, prin această pierdere a achizițiilor anterioare. Nici copiii cu autism, nici cei cu tulburare Asperger nu pierd achizițiile ci fac progrese psihomotorii menţinându-şi acel deficit de comunicare şi relaționare. Copiii cu Sindrom Rett suferă un proces de degradare neurologică, cu ataxia trunchiului, pierderea funcției de prehensiune a mâinilor; Se impune diagnostic diferențial şi cu Sindromul Landau-Kleffner (Afazie cu epilepsie) copii care au achiziționat limbajul dar treptat îl pierd însă copiii cu tulburare dezintegrativa nu au crize epileptice.
Medicii diagnostichează clinic sindromul Rett prin observarea semnelor și simptomelor în timpul creșterii și dezvoltării timpurii a copilului și prin efectuarea evaluărilor continue ale stării fizice și neurologice a copilului. Oamenii de știință au dezvoltat un test genetic pentru a completa diagnosticul clinic, care presupune căutarea mutației MECP2 pe cromozomul X al copilului.
Diagnosticul diferențial trebuie efectuat cu toate bolile metabolice care pot prezenta acest sindrom deteriorativ; prezenţa malformațiilor, a întârzierii în dezvoltarea psihomotorie din primii ani de viaţă, dar mai ales investigațiile metabolice ne ajută la diferențierea de tulburarea dezintegrativă, în care copiii sunt aparent normali în primii 2 - 3 ani de viaţă.
Condiția este genetică dar nu ereditară: se poate transmite în orice familie, în orice moment. Persoanele cu sindromul Rett suferă de dizabilități fizice, comunicare şi de învăţare accentuate şi sunt total dependenți de alții, necesitând sprijinul celorlalţi pe tot parcursul vieții lor. Cei mai mulți dintre bolnavi nu vor putea verbaliza şi doar 50% dintre aceștia vor putea merge, până la maturitate. Regresul sau întârzierea în dezvoltare apar începând de la 6 la 18 luni.
Sindromul Rett este cauzat de mutații pe cromozomul X al unei gene numite MECP2. Există mai mult de 200 de mutații diferite care pot fi găsite la nivelul genei MECP2. În funcţie de locul mutației de la nivelul genei, simptomele bolii sunt diferite de la o persoană la altă.
Severitatea sindromului este dificil de constatat şi poate varia mult de la debilitate gravă la ușoară, cu grade variabile de independentă. Simptomele şi gravitatea lor sunt diferite şi se pot modifica în timp. Nu toți oamenii vor avea aceleași simptome şi efectele la terapii pot fi diferite.
În prezent nu există niciun tratament pentru această afecțiune, dar există multiple tratamente care pot ameliora simptomele tulburării.
Tratament
Deși nu există un tratament pentru sindromul Rett, tratamentele sunt orientate pe simptome și spre oferirea de sprijin, ceea ce poate îmbunătăți potențialul de mișcare, comunicare și participare socială.
O abordare multidisciplinară în echipă este folosită de obicei pentru a trata persoana pe tot parcursul vieții. Această echipă poate include un medic de îngrijire primară, un fizioterapeut, un terapeut ocupațional, un logoped, un nutriționist și servicii de sprijin în mediul academic și ocupațional. Nevoia de tratament și de sprijin nu se termină odată cu înaintarea în vârstă a copiilor - de obicei este necesară pe tot parcursul vieții. Tratarea sindromului Rett necesită o abordare în echipă.
Tratamentele care pot ajuta copiii și adulții cu sindromul Rett includ:
Îngrijire medicală regulată. Gestionarea simptomelor și a problemelor de sănătate poate necesita o echipă multidisciplinară. Este necesară monitorizarea regulată a modificărilor fizice, cum ar fi scolioza și problemele gastrointestinale și cardiace.
Medicamente. Deși medicamentele nu pot vindeca sindromul Rett, acestea pot ajuta la controlul unor semne și simptome asociate cu această tulburare, cum ar fi crizele, rigiditatea musculară sau probleme cu respirația, somnul, tractul gastrointestinal sau inima.
Terapia fizică. Kinetoterapia și utilizarea de orteze sau ghipsuri pot ajuta copiii care au scolioză sau care au nevoie de sprijin pentru mâini sau articulații. În unele cazuri, fizioterapia poate ajuta, de asemenea, la menținerea mișcării, la crearea unei poziții de ședere corespunzătoare și la îmbunătățirea abilităților de mers, a echilibrului și a flexibilității. Dispozitivele de asistență pot fi utile.
Terapia ocupațională. Terapia ocupațională poate îmbunătăți utilizarea intenționată a mâinilor pentru activități precum îmbrăcarea și hrănirea. Dacă mișcările repetitive ale brațelor și mâinilor reprezintă o problemă, pot fi utile atelele care restricționează mișcarea cotului sau a încheieturii mâinii.
Logopedia. Terapia logopedică poate contribui la îmbunătățirea vieții unui copil prin învățarea modalităților nonverbale de comunicare și prin ajutor în interacțiunea socială.
Sprijin nutrițional. O nutriție adecvată este extrem de importantă pentru o creștere normală și pentru îmbunătățirea abilităților mentale, fizice și sociale. Poate fi recomandată o dietă bogată în calorii și bine echilibrată. Strategiile de hrănire pentru a preveni sufocarea sau vărsăturile sunt importante. Unii copii și adulți pot avea nevoie să fie hrăniți prin intermediul unui tub plasat direct în stomac (Gastrostoma).
Intervenție comportamentală. Practicarea și dezvoltarea unor bune obiceiuri de somn pot fi utile pentru tulburările de somn.
Servicii de sprijin. Formarea de abilitați academice, sociale și profesionale pot ajuta la integrarea în școală, la locul de muncă și în activitățile sociale. Adaptările incluzive pot face posibilă participarea într-o gama larga de activităţi.
| Share pe Facebook |